


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Atypical Teratoid/Rhabdoid Tumor Mimicking Tubercular Meningitis in a Child
Geetanjali S.Rathore,Sanath K.Reddy,Peter Abasolo,Paul D.Larsen
2.Department of Neurological Sciences, 988440 Nebraska Medical Center Omaha, NE 68198-8440.
3.Department of Pathology, Children's Hospital & Medical Center 8200 Dodge St Omaha NE 68114.
4.Pediatric Neurology, Department of Pediatrics 982163 Nebraska Medical Ctr Omaha, NE 68198, USA.
Copyright : © 2018 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Leptomeningeal enhancement in children without a mass lesion usually leads to a diagnosis of infectious meningitis. Atypical teratoid/rhabdoid tumor (AT/RT) is a rare, highly malignant central nervous system tumor typically presenting as an expanding mass, with secondary leptomeningeal involvement. We report a 19month old girl presenting with encephalopathy, opisthotonus and left facial palsy. MRI brain with contrast showed leptomeningeal enhancement of suprasellar cisterns and multiple cranial nerves bilaterally, without evidence of a definite mass. MRI spine showed diffuse smooth enhancement of the cauda equina with a small enhancing extradural lesion at T7. Cerebrospinal fluid studies (CSF) showed pleocytosis (monocytic predominance), markedly elevated protein and undetectable glucose. Cytology showed no malignant cells. She was initially misdiagnosed with tubercular meningitis, but showed no improvement on antitubercular therapy. An extensive infectious work up, including tuberculosis and fungal infections, remained negative. A brain biopsy led to the final diagnosis of ATRT. In conclusion, even though infectious meningitis is the most likely etiology of leptomeningeal enhancement in pediatrics, an atypical presentation should raise a concern for possible neoplastic process. Diffuse leptomeningeal features and cranial neuropathies should prompt an investigation for AT/RT as a possible cause especially in young children.
ATRT, leptomeningeal enhancement, basilar meningitis, tubercular meningitis,Neuroscience
1. Introduction
Atypical teratoid/rhabdoid tumor (ATRT) is a rare, highly malignant central nervous system tumor typically presenting as an expanding mass, with secondary leptomeningeal involvement [1]. It usually presents in young children less than 3 years old, even though reported in adults too [1,2]. Leptomeningeal enhancement in children without a mass lesion, with normal blood counts, usually leads to a diagnosis of infectious meningitis. We report here the case of a toddler presenting with encephalopathy, dystonia and multiple cranial nerve deficits. She was initially misdiagnosed with tubercular meningitis based on imaging and spinal fluid studies. She showed no improvement on antitubercular therapy and an extensive infectious workup was negative, prompting a brain biopsy which led to the final diagnosis of AT/RT. The clinical presentation of AT/RT can be variable and imaging does not always help distinguish it from other CNS tumors and infections. Histologic, immunohistochemical, and cytogenetic studies are typically needed to make the diagnosis. The loss of INI1 expression by immunohistochemical staining and detecting SMARCB1 deletions via FISH studies helps confirm the diagnosis of AT/RT [2,3]. The long term survival rates are poor even with aggressive multimodal therapies [4,5].
2. Clinical Summary
A nineteenmonth old Caucasian female presented with a two week history of vomiting without fever or diarrhea. Over the next few days she got progressively less active and somnolent, with episodes of back arching.
Initial MRI of the brain without contrast wasnormal. CSF showed WBC of 20 with monocytic predominance, Protein was 88 and Glucose was undetectable. She was found to have Ecoli UTI so treated withceftriaxone.
No significant clinical improvement was seen over the next week so MRI brain was repeated with contrast. This showed diffuse leptomeningeal enhancement of right suprasellar cistern, right sylvian fissure, right temporal and frontal lobes along with contrast enhancement of bilateral optic nerves, chiasm, radiation, bilateral trigeminal, left facial and vestibulo-cochlear nerves. Right basal ganglia contrast enhancement along with diffusion restriction was also noted (Figure 1 A, B, C)
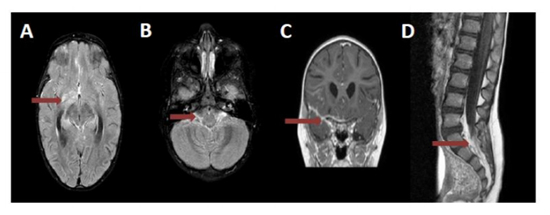
Repeat CSF testing a week after initial studies showed an increase in WBC from 20 to 56 and protein increased from 88 to 118, CSF Glucose remained undetectable.
Father had recently traveled to Singapore and Dubai but asymptomatic. Paternal grandmother had a chronic cough with recent travel to Italy and the Mediterranean. The patient was healthy with no significant past medical history besides atraumatic non displaced skull fracture 6 months ago. Based on imaging/CSF findings and potential exposure, a presumptive diagnosis of tubercular meningitis was made and she was started on empiric anti-tubercular treatment with Rifampin, Isoniazid, Pyrazinamide and Ethionamide along with B6 supplementation. Dexamethasone was started to reduce inflammation.
Our patient continued to progressively get worse on antibiotics. Her Neurological exam showed right sided relative afferent pupillary defect (RAPD), left facial palsy, brisk symmetrical reflexes, and good strength. She was able to walk independently but with neck stiffness and opisthotonic posturing of the lumbar area. Normal funduscopic and retinal exam noted by ophthalmology.
3. Differential Diagnosis And Further Workup
Her PPD (purified protein derivative) test and mycobacterium tuberculosis PCR on CSF were both negative. Quantiferongold was negative for TB. Chest X-ray was normal. She was also tested negative for HIV, Mycoplasma, Cryptococcus, Histoplasma, Lyme, syphilis, Lympho-Chorio-Meniningitis virus, Leptospira, Brucella, Toxoplasma and EBV. Broad range PCR for bacteria, fungi and AFB was negative. ACE (angiotensin converting enzyme) level in serum and CSF was not elevated, ruling out sarcoidosis. Anti NMDA receptor antibody testing and autoimmune encephalitis panel was negative. CSF oligoclonal bands were absent, myelin basic protein was within normal range. IgG synthesis rate was elevated at 53.7 with an highly elevated CSF albumin at 280, Elevated albumin index at 77, all indicating a moderate to severe breakdown of blood-brain barrier As there was worsening of lumbar spine lordosis with arching, MRI of the brain along with MRI of the entire spine was repeated. This showed stable lesions in the brain, but new diffuse cauda equina enhancement (Figure 1D). Repeat lumbar puncture showed increase in WBC to 106, increase in protein to greater than 600 with albumin predominance on CSF electrophoresis and a normal serum electrophoresis. CSF cytology showed atypical cells with occasional pyknotic nuclei and mitotic figures. Flow cytometric studies demonstrated CD3 positive T-cells with monocytic macrophages; B-cells were polyclonal by light chain distribution. No blasts were identified.
No clear etiology was determined after an extensive workup, therefore neurosurgery was consulted for a brain biopsy. The right temporal area grossly appeared as a firm, vascularized and abnormal-appearing blanched tissue in pia and subpia of temporal tip. Whitish-appearing collection was present beneath arachnoid in multiple sulci near the sylvian fissure.
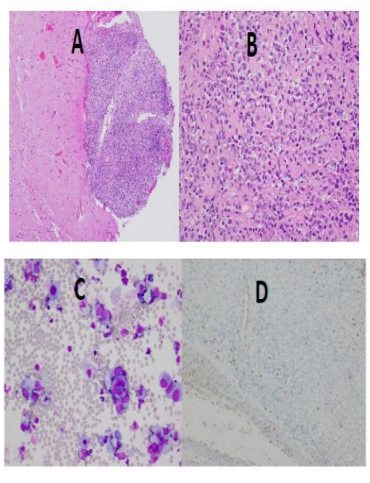
The H&E sections showed a malignant tumor involving the meningeal surface with focal extension into the glial tissue (Figure 2A). The malignant cells showed variable cohesiveness and were medium to large with increased nuclear to cytoplasmic ratios and eccentric, large nuclei with irregular contours, and occasional nucleoli (Figure 2B and 2C). Immunohistochemical stains were performed; the malignant cells were positive for vimentin and showed focal positivity with cytokeratin and epithelial membrane antigen. These cells were negative for CD45 and CD20. A high Ki-67 index was observed. The tumor did not retain INI1 nuclear staining (Figure 2D). Loss of SMARCB1 (INI1; 22q11.23) expression was detected by FISH. The morphologic, immunophenotypic and FISH studies were all supportive of the diagnosis of an atypical teratoid/ rhabdoid tumor (AT/RT).
CT of the chest/abdomen, pelvis showed no evidence of malignant lesions. Anti-tubercular therapy was stopped and she was started on chemotherapy according to the Dana Farber AT/RT protocol. Due to significant side effects of intensive chemotherapy without much improvement, family decided to proceed to palliative care.
B) & C) Higher resolution H&E showing variable cohesiveness, increased nuclear to cytoplasmic ratios and eccentric, large nuclei with irregular contours
D) Absence of INI 1 nuclear staining
4. Discussion
Atypical teratoid/ rhabdoidtumor (AT/RT) is a rare, highly malignant CNS tumor primarily occurring in children less than 3 years old. It comprises of 1-2% of all pediatric brain tumors [1] most commonly these tumors occur infra-tentorially, with about two-thirds arising from the cerebellum [2]. They have however been reported in supra-tentorial locations including the cerebral hemispheres, pineal gland region, septum pellucidum and hypothalamus [2]. Up to 30% of tumors have disseminated at diagnosis, with leptomeningeal spread being the most common[3].
There are no specific imaging features for diagnosis of intracranial AT/RT [3]. AT/RT tends to be hyperdense on CT and enhances intensely. Typically present as large heterogenous masses often with some component of calcification, hemorrhage or necrosis. Nodular, clumpy enhancement of the leptomeninges, along the cord and into the cauda equina may be seen given the high tendency for disseminated leptomeningeal disease [4]. Our patient had an atypical presentation with a primary leptomeningeal AT/RT, without evidence of a solid intra-parenchymal mass. Such a presentation has not been reported in the literature, thus making this rare diagnosis even more challenging.
The clinical presentation and imaging studies do not definitively distinguish AT/RT from other CNS tumors and infections, especially if given an atypical presentation. Histologic, immunohistochemical, and cytogenetic studies helpconfirm the diagnosis [2]. A tumor with histologic features including a loose, disordered, complex architecture with pleomorphic large malignant cells showing eosinophilic cytoplasm, large eccentric nuclei, and nucleoli would raise concern for ATRT. Immunohisto chemical studies for epithelial membrane antigen, vimentin, and smooth muscle actinwould typically be immunoreactive in an AT/RT, while a PNET would be negative for those markers [2]. Bi-allelic inactivation of the SMARCB1 (INI1/SNF5/BAF47) gene on the long arm of chromosome 22 is noted in a majority of ATRT (SMARCATRT). In addition to observing loss of INI1 expression by immunohistochemical stain, SMARCB1 FISH studieshelp to confirm the diagnosis by detecting SMARCB1 deletions[3,4], as seen in our patient.
With an aggressive nature of AT/RT median survival rates are reported to be only 6 to 17 months. Younger age and leptomeningeal spread heralding poorer prognosis [5,6]. Various treatment modalities, including multi-agent chemotherapy, intrathecal chemotherapy and radiotherapy have been utilized for children with AT/RT, the results however remain suboptimal with not much improvement in survival rate [4]. High-dose chemotherapy followed by autologous stem cell rescue (HD-SCR) has been used to consolidate initial chemotherapy in order to try to delay use of radiation therapy. This has shown to have improved survival in newly diagnosed patients with AT/RT in a small case series [5]. With better understanding of the molecular biology of AT/RT new drug trials are in process targeting the modulators of hSNF5/INI1 gene 1 (insulin like growth factor, cyclin D, Aurora A and tyrosine kinases) in hope to discover potential therapeutic targets [7].
5. Conclusion
AT/RT is a very rare neoplasm and with a diverse presentation. Even though infection is the most common cause of leptomeningeal enhancement without a primary solid mass in the pediatric population, neoplasm should remain on differential. Basilar enhancement with multiple cranial nerve involvement should make neoplasm higher on the differential. As imaging and spinal fluid analysis may not lead to diagnosis in most cases, tissue for immunohistology should be obtained to establish diagnosis.
References
- Y. T. Udaka, K. Shayan, N. A. Chuang, and J. R. Crawford; Atypical presentation of atypical teratoidrhabdoid tumor in a child: Case Rep Oncol Med. 2013; 2013:815923. doi: 10.1155/2013/815923. Epub 2013 May 27.
- MeenakshiBhattacharjee, John Hicks, Lauren Langford, Robert Dauser, Douglas Strother, MuraliChintagumpala, Marc Horowitz, Linda Cooley & Hannes Vogel (1997) Central Nervous System Atypical Teratoid/Rhabdoid Tumors of Infancy and Childhood, Ultrastructural Pathology, 21:4, 369-378, DOI: 10.3109/01913129709021935
- Kong Jung Au Yong & Jacob L. Jaremko& Lennart Jans& Ravi Bhargava & Lee T. Coleman &Vivek Mehta & Michael R. Ditchfield; How specific is the MRI appearance of supratentorial atypical teratoidrhabdoid tumors? PediatrRadiol (2013) 43:347-354; DOI 10.1007/s00247-012-2530-z
- Wells, Elizabeth M. MD; Packer, Roger J. MD, FAAN: Pediatric brain tumors: CONTINUUM: Lifelong Learning in Neurology: April 2015 - Volume 21 - Issue 2, Neuro-oncology - p 373- 396, doi: 10.1212/01.CON.0000464176.96311. d1
- Schrey D, CarcellerLechon F, Malietzis G, Moreno L, Dufour C, Chi S, Lafay-Cousin L, von Hoff K, Athanasiou T, Marshall LV, Zacharoulis S: Multimodal therapy in children and adolescents with newly diagnosed atypical teratoidrhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol. 2016 Jan;126(1):81-90. doi: 10.1007/s11060-015-1904-0. Epub 2015 Nov 25.
- NilgunSelcuk, Murat Elevli, DicleInanc, HuseyinArslan; Atypical Teratoid/Rhabdoid Tumor Mimicking Tuberculous Meningitis, Indian Pediatr 45 (4), 325-326. 4 2008.
- Ginn KF, Gajjar A; Atypicalteratoidrhabdoid tumor: current therapy and future directions. Front Oncol. 2012 Sep 12; 2:114. doi: 10.3389/fonc.2012.00114. eCollection 2012.